The cystatin C or γ trace is a group of proteinase inhibitors and is present in the body fluids of all mammals. In vitro experiments have confirmed cystatin has a capacity of regulating the activity of the cysteine protease (plays important role in intracellular catabolism of peptides and proteins). Cystatin C regulates the activity of cysteine proteases by inhibiting their activity. They are mainly active upon cysteine proteases like cathepsins B, H, K, L and S. Moreover, cystatin C is proteolytically degraded by cathepsin D and elastase. Cathepsins are lysosomal proteases commonly known as housekeeping genes and they vary in their substrate-specificities, structure and biochemical functions. The uncontrolled proteolysis of cystatin C due to imbalance in active proteases and their endogenous inhibitors lead to the generation of several debilitating diseases such as multiple sclerosis, muscular dystrophy, Alzheimer disease (AD), rheumatoid arthritis, ischemia, renal failure, osteoporosis, inflammatory disease and various other types of cancer (1).
Cerebral Amyloid Angiopathy (CAA) is an autosomal dominat disease. It is a combination of amyloidosis with hereditary cerebral hemorrhage and is commonly known as hereditary cystatin C amyloid angiopathy (HCCAA). In this diseases, the amyloid deposition in the cerebral/ spinal arteries and arterioles leads to the generation of recurrent hemorrhagic strokes generating severe brain damage, leading to fatal stroke. Leu68Gln variant of cystatin C gene is the principal component of the amyloid fibrils. A heterozygous point mutation, in the cystatin C gene is responsible for this CAA autosomal dominant disease. Such heterogeneous point mutation results in a truncated version of the cystatin c gene from the amio terminal domain. The truncated protein is not being recognized by the elastase or a serine protease and this lead ot protein misfolding and subsequent deposition over the cerebral/spinal arteries as amyloid fibrils (2).
CST3 is commonly known as human cystatin C gene. It is located in chromosome 20 and its mutation is responsible for hereditary cystatin C amyloid angiopathy (3).Affect of Cystatin C-Cerebral Amyloid Angiopathy on patients, medicine and society
CAA is regarded as the morphologic hallmarks of Alzheimer disease (AD). However, its presence has been detected in the CT scan of elderly patients who are neurologically healthy. While majorly asymptomatic, CAA may often lead to intracranial hemorrhage (ICH) and dementia. Moreover, ICH is the most recognized result of CAA.
It has a profuse impact on the society, mostly over the senior citizens and thus impacting the elderly care program. Magnetic Resonance Imaging (MRI) is one the principal technique used for the detection of the CAA (1).
Human Cystatin C Gene or CST3 is found in mammals. It belongs to a group of proteinase inhibitors and regulate the mode of action of cysteine proteinases by inhibiting their activity (4).
It is located in chromosome number 20 in humans (4).
CST3 gene is composed of three exons and two intron sequences. The introns has a sequence of 2252 and 1254 bp respectively. The resulting gene size is 4.3 kb approx. The introns are located in between the nucleotide triplets encoding amino acid residues 55-56 and 93-94 of the mature protein respectively. The respective positions of the introns are identical with cystatin SN and cystatin SA genes. However, the first intron of CST3 is comparatively bigger than the first introns present in the cystatin SN and cystatin SA genes (4).
The CST3 is expressed throughout the body fluid and seminal plasma detected the highest concentration of CST3 genes. The body fluid circulated throughout the body and deliver CST3 uniformly. However, the major infiltration is seen in liver, kidney, small intestine, antrum, stomach, lung, placenta and seminal vesicle. There are certain non-tissue specific expression of CST3 and this is attributed to the its 5′-flanking region. It shares identical properties with the promoter domain of ‘housekeeping genes.
Three cystatin loci namely CST1 (cystatin SN), CST2 (cystatin SA) and CSTP1 (a cystatin pseudogene) together constitute CST3 (5).
The expression of CST3 is increase in the facial nucleus under the influence of the several pathological conditions and majorly in axotomy. Other predominant cell type responsible CST3 expression is microglia and not astrocyte or neuron. The up-regulation of cystatin C by microglia or hyperplasia of microglia to be presice leads to the expression of the CST3. Microglia significantly proliferate in during brain injuries such as axotomy or stab wound and this lead to the overall expression of the CST3 gene. On the other hand, it can also be stated that CST3 controls the turn-over of intracellular cytosolic proteins of microglia (6).
Mutation in the amino terminal domain of the CST3 gene results in the protein misfolding during the post translational modification. Such misfolding results in the deposition of the CST protein in the form of amyloid fibrils in the brain and neurons. CST3 is also foun97d being codeposited in the amyloid plaques of Alzheimer’s disease or Down’s syndrome (7).
The CST3 gene of mammals shows structural similarity with the Cystatin C gene of the mice. Such animal model has been extensively utilized in research for the study of the disease prognosis. It is used as a disease model for Alzheimer’s disease, marker for renal function affected by cystatin C and as a cerebral hemorrhage model (8).
Normal Protein Function And Role In Human Biology
The cystatin C or γ trace is a group of proteinase inhibitors. It is widely distributed inside the body fluids of all mammals and is ubiquitous in nature. Cystatin C regulates the activity of the cysteine protease, which plays an important role in intracellular catabolism of peptides and proteins. Cystatin C mainly regulates the activity of cysteine proteases via inhibiting their function. They are mainly active upon cysteine proteases like cathepsins B, H, K, L and S. Moreover, cystatin C undergoes proteolytic degradation by cathepsin D and elastase. Cathepsins are lysosomal proteases and are popularly known as housekeeping genes and they differ in their primary structural symmetry, substrate binding specificities and biochemical characteristics.
Cystatin C or cystatin C gene to be precise is widely used as a biomarker of normal renal function. It also plays a major role in the onset or in the deterioration of the cardiovascular disease
Cystatin C belongs to the cystatin superfamily. It interacts reversibly with target peptidases and such interactions is seemingly independent and depends only on the affinity contributions from a wedge-shaped binding region that is constructed with the help of the two loop-forming inhibitor segments and a corresponding binding region corresponding to the N-terminal segment of that particular inhibitor.
It is composed of 120-residue of non-glycosylated polypeptide chain. The cystatin C gene exists as a single copy of 5 kb (approx) in the human genome The amino-acid sequences of low-molecular weight in the cystatin family have significant homology with each other. However, the post-translational modification of each family of cystatin gene (alpha, beta and gamma) is different from each other. Such different approach in the post translational modification depends on their localizations and different biological functions (4).
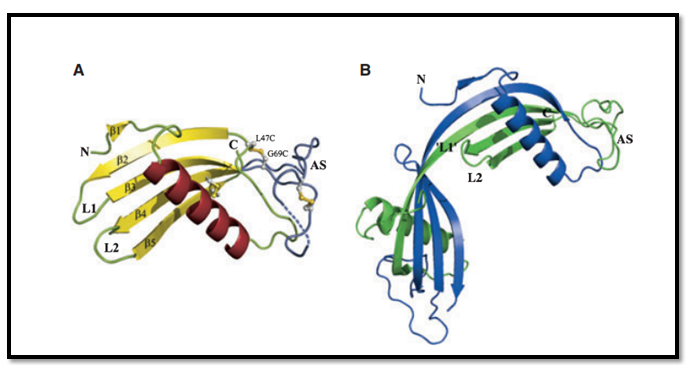
Secondary Tertiary And Quaternary Structural Properties
Figure: 3D structure of Cystatin protein
(Source: 9)
The two Cystatin molecules (as displayed in picture A and B) has canonical cystatin fold, (N)–b1– a–b2–L1–b3–AS–b4–L2–b5–(C), with a five-stranded antiparallel b-sheet gripped around a long alpha helix. The appending structure (AS) is broad and irregular. It is positioned at the opposite end of the beta sheet relative to the N-terminus⁄ loop L1 ⁄ loop L2 edge. Such specialized structural symmetry is known as papain-binding epitope. b1 is the one of the shortest elements out of the rest standard five antiparallel beta platted sheet. It comprises of two residues. In both the molecules displayed in the picture, the first 11 residues are completely disordered. The following two AS residues, which differ in each molecule are found to be Proline78-Leucine79 in A and Leucine80-Aspartate81 in B. Some of the disulfide bridges exists in a partially broken form in the tertiary structure (9).
The mutation in the one of the three exons of cystatin protein results in the occurance of the CAA disease. This mutation occurs in the luciene residue causes at 68th position in the gene. Leu68Gln variant of cystatin C gene is the principal component of the amyloid fibrils. A heterozygous point mutation, in the cystatin C gene is responsible for this CAA autosomal dominant disease. Such heterogeneous point mutation results in a truncated version of the cystatin c gene from the amio terminal domain. The truncated protein is not being recognized by the elastase or a serine protease and this lead ot protein misfolding and subsequent deposition over the cerebral/spinal arteries as amyloid fibrils (10).
Three cystatin loci namely CST1 (cystatin SN), CST2 (cystatin SA) and CSTP1 (a cystatin pseudogene) together constitute CST3 (5)(11).
There are extensive biomedical research that has been carried on Cystatin in order to ascertain the actual reason and the principal stigma behind the prognosis of CAA, a disease prevalent in elderly people. In order to study the effect and the intensity of the mutation and subsequent protein misfolding on the disease prognosis, several animal model, mainly mouse have been designed and studies extensively.
Though significant studies have been undertaken in this field, but keeping the complexity of the disease in mind, further through research in the field centering the point specific mutation of CST3 gene and its effects on CAA is required (12)
Delivering a high-quality product at a reasonable price is not enough anymore.
That’s why we have developed 5 beneficial guarantees that will make your experience with our service enjoyable, easy, and safe.
You have to be 100% sure of the quality of your product to give a money-back guarantee. This describes us perfectly. Make sure that this guarantee is totally transparent.
Read moreEach paper is composed from scratch, according to your instructions. It is then checked by our plagiarism-detection software. There is no gap where plagiarism could squeeze in.
Read moreThanks to our free revisions, there is no way for you to be unsatisfied. We will work on your paper until you are completely happy with the result.
Read moreYour email is safe, as we store it according to international data protection rules. Your bank details are secure, as we use only reliable payment systems.
Read moreBy sending us your money, you buy the service we provide. Check out our terms and conditions if you prefer business talks to be laid out in official language.
Read more